

{kind=link}
TRATAMENTO DA SINDROME DE CROUZON
A síndrome de Crouzon faz parte das chamadas craniossinostoses sindrômicas, que constituem-se de um conjunto de malformações de origem genética que apresentam como característica comum a fusão (ou sinostose) precoce de uma ou, geralmente, múltiplas suturas cranianas, inibição do crescimento das sincondroses da base do crânio e sinostose das suturas dos ossos cranianos e faciais, particularmente entre a maxila e o esfenóide (Aduss 1981). Tal situação provoca um desbalanço entre o crescimento ósseo (craniano) e de partes moles (encefálico), levando por vezes ao surgimento de hipertensão intracraniana (HIC) pela restrição do crescimento cerebral, que pode acarretar em prejuízo no desenvolvimento cognitivo destes pacientes (Gabarra 2000).
De transmissão autossômica dominante, é conseqüência de uma mutação no gene responsável pela codificação dos receptores do fator de crescimento fibroblástico tipo 2 (FGFR2), localizado no braço longo do cromossomo 10 (10q26) (Kan et al 2002 e Chang et al 2006). Sua incidência varia entre 15,5 e 16,5 por milhão de nascimentos (Cohen Junior 1992).
Foi descrita originalmente por Octave Crouzon em 1912, que descreveu a tríade clássica de alterações cranianas (notadamente a braquicefalia), exorbitismo e hipoplasia da face média (Crouzon 1912). Além dessas características, os pacientes podem também apresentar hipertelorismo, estrabismo, alterações nasais, diminuição do espaço nasofaríngeo, diminuição do comprimento da fossa craniana anterior com elevação das asas menores do esfenóide, lâmina cribiforme deprimida, aumento no tamanho dos seios etmoidais, palato em ogiva e dentes aglomerados (Aduss 1981).
Diversos fatores são capazes de influenciar o desenvolvimento cognitivo de pacientes com craniossinostoses sindrômicas. Renier et al (1996) mostraram que, em pacientes com de síndrome de Apert, também foram importantes alterações morfológicas encefálicas (anomalias do septo pelúcido) e a qualidade do ambiente familiar (39,3% dos pacientes com estrutura familiar normal alcançaram um QI normal, enquanto somente 12,5% dos pacientes institucionalizados alcançaram este limite). Ainda tratando-se da síndrome de Apert, Yacubian-Fernandes et al (2005) encontraram um índice de deficiência cognitiva de 28,5% (4 pacientes de uma amostra de 18), sendo que a condição sócio-econômica das famílias e o nível de instrução dos pais também foram citados como sendo fatores relevantes no desenvolvimento cognitivo destes pacientes. Estudos detectaram alterações encefálicas em 56% desta população com síndrome de Apert (Yacubian-Fernandes et al 2004).
Entretanto, apesar de haver diversos estudos tentando correlacionar fatores que possam interferir no desenvolvimento neuropsicológico na síndrome de Apert, existem poucos trabalhos na literatura específicos para a síndrome de Crouzon, possivelmente porque o índice de retardo mental neste grupo é muito menor quando comparado ao Apert (David et al 1982), e com uma prevalência de alterações estruturais cerebrais também menor. Aguado-Balsas et al (1999) realizaram uma revisão da literatura e concluíram que a deficiência cognitiva nesta síndrome é leve e irregular, apresentando comprometimento de certas faculdades mentais de forma variada. Noetzel et al (1985) citaram que a incidência de deficiência cognitiva na síndrome de Crouzon pode chegar a 20%, enquanto que em craniossinostoses monossuturais varia de 6,9% a 9,5%.
Postularam ainda que, entre as causas de retardo mental nas craniossinostoses sindrômicas, as principais seriam: A) hidrocefalia com aumento da PIC e destruição progressiva dos neurônios; B) causas não relacionadas diretamente à craniossinostose, como prematuridade, infecções ou trauma; e C) anormalidades embriogênicas afetando o encéfalo, confirmadas pela presença de ventriculomegalias não progressivas em muitos pacientes deste grupo.
A aquisição e o desenvolvimento da linguagem também dependem de vários fatores: condições neurológicas ideais (integridade anatômica e funcional do sistema nervoso central e periférico), aparelho fonatório adequado, condições sociais, afetivas (qualidade do estímulo) e cognitivas. Alterações da linguagem são problemas freqüentes do desenvolvimento na infância, atingindo de 3 a 15% das crianças (Meirelles et al 2006).
As craniossinostoses podem ser prejudiciais considerando o processo de aquisição e desenvolvimento de linguagem, pois atingem estruturas orofaciais, comprometem o sistema auditivo, interferem no desenvolvimento neuropsicológico e também dificultam a adaptação social da criança (Meirelles et al 2006).
Contudo, também são escassos os estudos avaliando o desenvolvimento da linguagem nos pacientes com síndrome de Crouzon. Durante muito tempo, existiram somente descrições empíricas e pontuais a respeito das habilidades de linguagem em pacientes portadores de craniossinostoses sindrômicas, devido principalmente a dois fatores: a baixa incidência de tais patologias, que proporciona casuísticas pequenas, e a gravidade das malformações, freqüentemente tão importante a ponto de se relegar os aspectos relacionados à avaliação de linguagem e comunicação a segundo plano (Peterson 1973). Este autor relatou sua experiência pessoal com 5 casos de síndrome de Crouzon, descrevendo desde linguagem normal até alterações leves. Em uma revisão mais abrangente, chama a atenção para a estreita relação entre os aspectos psicológicos e as habilidades de comunicação.
Tratamento
O tratamento desta patologia é bastante complexo, envolvendo diversas opções terapêuticas em diferentes fases de evolução dos pacientes, e consiste sempre na participação de uma equipe multidisciplinar (Turvey et al 1979), com a integração de pediatras, cirurgiões plásticos, neurocirurgiões, enfermeiros, fonoaudiólogos, assistentes sociais, psicólogos, dentistas e cirurgiões buco-maxilo-faciais, entre outros.
Do ponto de vista do tratamento cirúrgico, divide-se em duas fases: uma primeira fase precoce, a ser realizada idealmente no primeiro ano de vida, no intuito de promover uma expansão craniana e evitar as conseqüências deletérias da HIC, além de garantir uma melhor proteção ocular diminuindo a exposição dos globos oculares e melhorando o aspecto do exorbitismo. A segunda fase ocorre idealmente após a erupção da dentição definitiva, com o objetivo de realizar o avanço da face média, seja cirurgicamente (através de fratura tipo Le Fort III), seja através de tração esquelética por distratores (Aduss 1981).
Frente a todas as intercorrências e patologias associadas que os pacientes com síndrome de Crouzon podem apresentar, bem como às diferentes necessidades terapêuticas em diferentes estágios do desenvolvimento, o tratamento multidisciplinar impõe-se como o ideal (Turvey et al 1979).
No intuito de evitar os efeitos deletérios da HIC no desenvolvimento cerebral, preconiza-se a cirurgia precoce de remodelação craniana para as cranioestenoses sindrômicas como Crouzon e Apert.
Inicialmente as técnicas neurocirúrgicas desenvolvidas no início do século 20 consistiam em craniectomias em faixa (ou suturectomias) sobre as suturas fechadas, esperando que o crânio, uma vez descomprimido, se remodelasse e continuasse a crescer de forma mais simétrica (Posnick e Ruiz 2000). Entretanto, tal técnica mostrou-se insuficiente para corrigir tais problemas.
Os grandes avanços nesse campo foram feitos por Paul Tessier na década de 70, quando demonstrou ser possível realizar o avanço frontal, orbitário e da face média num único procedimento, e também agregar a correção do hipertelorismo quando necessária (Aduss 1981). Desde então, o avanço fronto-orbitário tem se tornado o procedimento padrão para correção de tais malformações craniofaciais.
Alguns autores preconizam o momento entre 4 e 12 meses (Renier et al 1996) e outros consideram tempo ideal até 36 meses (Gabarra 2000). Vale a pena lembrar que o cérebro aumenta em peso cerca de 85% nos primeiros 6 meses de vida, chegando a 135% de aumento no final do primeiro ano. Apesar do crescimento tornar-se mais lento a partir desse momento, ele não cessará completamente até aproximadamente os 8 anos de idade (Aduss 1981). Posnick e Ruiz (2000) julgam que o tempo ideal para tais correções seja entre 9 e 11 meses, pois neste período o crânio e o rebordo orbitário mantêm-se melhor na posição corrigida.
Marchac e Renier (1996) dividiram o tratamento cirúrgico dos pacientes portadores de craniossinostoses sindrômicas em duas fases distintas: uma precoce (crânio) e outra tardia (face).
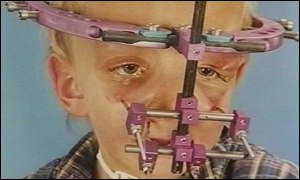
A primeira fase, precoce, a ser realizada idealmente no primeiro ano de vida, envolvendo a remodelação craniana, utilizando a técnica do avanço fronto-orbitário, com o objetivo de ganhar 2 centímetros anteriormente através do avanço do osso frontal e do rebordo orbitário, no intuito de conseguir uma boa proteção ocular e alívio efetivo da HIC (Marchac e Renier 1996). Recentemente, alguns autores têm preferido uma supercorreção, sobretudo quando a cirurgia é realizada muito precocemente (Posnick e Ruiz 2000).
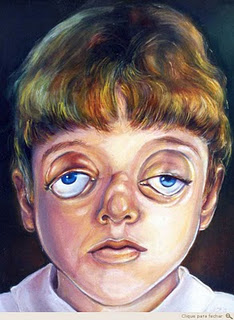
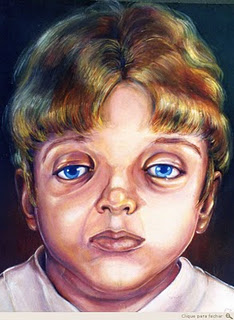
A segunda fase para correção das alterações faciais ocorre por volta dos 12 anos de idade (realizada através de fratura Le Fort III e avanço da face média), aguardando a erupção completa dos dentes definitivos. Dependendo das necessidades psicológicas da criança ou do grau de dificuldade respiratória, antecipa-se esta segunda fase (Mitsukawa et al 2004).
Postula-se que os pacientes com deformidades craniofaciais tratadas precocemente sofreriam menor trauma psicológico com relação à família e a sociedade em geral (McCarty et al 1984), devido a uma melhor remodelação craniana.